Axenfeld-Rieger Sendromu Nedir? Belirtileri, Nedenleri, Tedavisi
Axenfeld-Rieger sendromu (Axenfeld-Rieger syndrome), esasen bir göz bozukluğudur, ancak vücudun diğer kısımlarını da etkileyebilir.
Bu durum, ön segment olarak bilinen gözün ön kısmındaki anormalliklerle karakterizedir. Örneğin, gözün renkli kısmı (iris) ince veya yetersiz gelişmiş olabilir.
İris normalde gözbebeği adı verilen ve içinden ışığın göze girdiği tek bir merkezi deliğe sahiptir. Axenfeld-Rieger sendromlu kişilerde genellikle merkez dışı bir göz bebeği (korektopi) veya iriste birden fazla göz bebeği gibi görünebilen ekstra delikler (polikori) vardır. Bu durum ayrıca gözün açık ön kaplaması olan kornea anormalliklerine de neden olabilir.
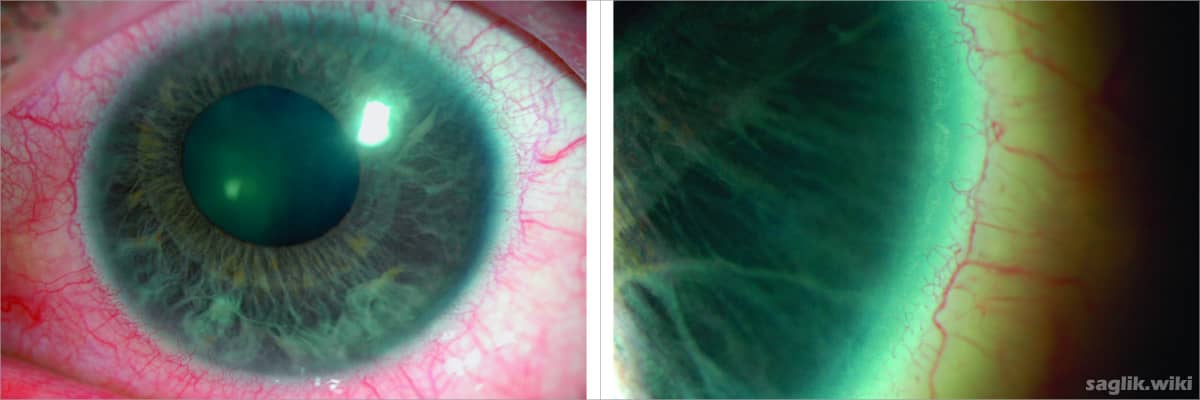
Etkilenen bireylerin yaklaşık yarısı, göz içindeki basıncı artıran ciddi bir durum olan glokom geliştirir. Glokom Axenfeld-Rieger sendromu ile ortaya çıktığında, bebeklik döneminde ortaya çıkabilmesine rağmen, çoğunlukla geç çocukluk veya ergenlik döneminde gelişir. Glokom görme kaybına veya körlüğe neden olabilir.
Diğer belirti ve semptomlar eşlik etmezse, göz anomalileri “Rieger göz anomalileri” olarak adlandırılır.
Axenfeld-Rieger sendromunun belirti ve semptomları vücudun diğer kısımlarını da etkileyebilir. Etkilenen birçok kişi, geniş aralıklı gözler (hipertelorizm), geniş ve düz bir burun köprüsü, basık bir orta yüz ve çıkık bir alın gibi ayırt edici yüz özelliklerine sahiptir.
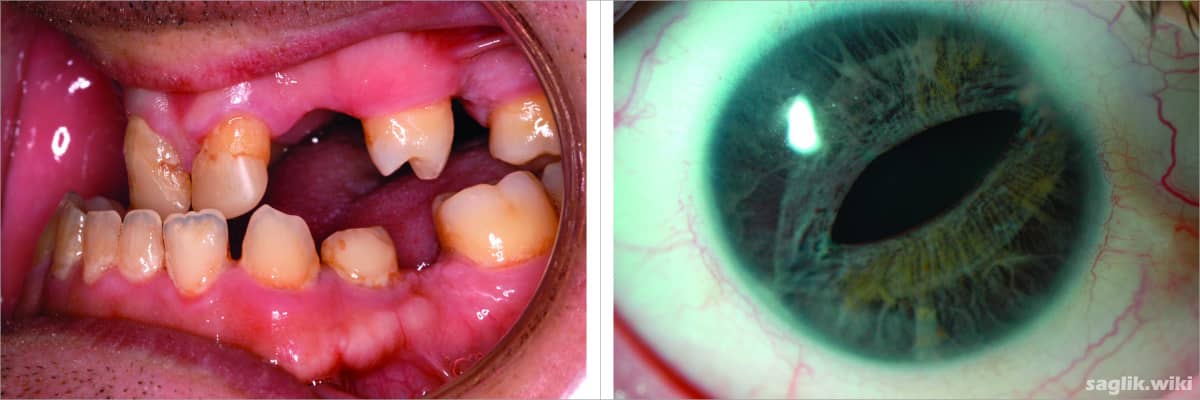
Bu durum aynı zamanda olağandışı küçük dişler (mikrodonti) veya normalden daha az diş (oligodonti) dahil olmak üzere diş anormallikleri ile de ilişkilidir. Axenfeld-Rieger sendromlu bazı insanlar göbek çevresinde ekstra deri kıvrımlarına sahiptir (fazlalık periumbilikal cilt).
Diğer, daha az yaygın özellikler arasında kalp kusurları, idrar deliğinin penisin alt tarafında açılması (hipospadias), anüsün daralması (anal stenoz) ve yavaş büyümeye neden olabilecek hipofiz bezi anormallikleri sayılabilir.
Araştırmacılar en az üç tip Axenfeld-Rieger sendromu tanımladılar. 1’den 3’e kadar numaralandırılan türler, genetik nedenleriyle ayırt edilir.
Axenfeld-Rieger Sendromu Ne Kadar Yaygındır?
Axenfeld-Rieger sendromu, erkekleri ve kadınları yaklaşık olarak eşit sayıda etkileyen nadir bir hastalıktır. Sendromun tahmini prevalansı (görülme oranı) 200.000 kişide 1’dir.
Axenfeld-Rieger Sendromunun Nedenleri
Axenfeld-Rieger sendromu, bilinen en az iki gendeki mutasyonlardan kaynaklanır, PITX2 ve FOXC1.
PITX2 gen mutasyonları tip 1’e ve FOXC1 gen mutasyonları tip 3’e neden olur. Tip 2 ile ilişkili gen muhtemelen 13. kromozomda bulunur, ancak tanımlanmamıştır.
PITX2 ve FOXC1 genlerinden üretilen proteinler, transkripsiyon faktörleridir, yani DNA’ya tutundukları (bağlandıkları) ve diğer genlerin aktivitesini kontrol etmeye yardımcı oldukları anlamına gelir.
Bu transkripsiyon faktörleri gelişmekte olan göz ve vücudun diğer bölgelerinde doğumdan önce aktiftir. Embriyonik gelişimde, özellikle gözün ön segmentindeki yapıların oluşumunda önemli rol oynadıkları görülmektedir.
PITX2 veya FOXC1 genindeki mutasyonlar, normal gelişim için gerekli olan diğer genlerin aktivitesini bozar. Bu genlerin bozulmuş regülasyonu (düzenlenmesi), gözün ön segmentinin ve vücudun diğer bölümlerinin oluşumunda sorunlara yol açar.
Bu gelişimsel anormallikler, Axenfeld-Rieger sendromunun karakteristik özelliklerinin temelini oluşturur. PITX2 gen mutasyonlarına sahip etkilenen bireylerin, FOXC1 gen mutasyonları olanlara göre, göz dışındaki vücut kısımlarını etkileyen anormalliklere sahip olma olasılığı daha yüksektir.
Axenfeld-Rieger sendromlu bazı kişilerde PITX2 veya FOXC1 genlerinde mutasyonlar tespit edilmemiştir. Bu bireylerde, durumun nedeni bilinmemektedir. Henüz tanımlanamayan diğer genler de Axenfeld-Rieger sendromuna neden olabilir.
Axenfeld-Rieger Sendromu Nasıl Kalıtılır?
Axenfeld-Rieger sendromu, otozomal dominant bir şekilde kalıtsaldır.
Vücudumuzdaki her genin iki kopyası var. Otozomal dominant koşullarda, bu genin sadece bir kopyasında bir mutasyon varsa, o kişi durumu geliştirecektir. Bu mutasyon, bir ebeveynden miras alınabilir veya o kişide ilk kez tesadüfen meydana gelebilir, buna “de novo” mutasyon denir.
Axenfeld-Rieger sendromlu bir bireyin her çocuğunun mutasyonu devralma şansı % 50’dir. Mutasyonu miras alan çocuklar, Axenfeld-Rieger sendromuna sahip olacak, ancak semptomları ebeveynlerinin semptomlarından daha fazla veya daha az şiddetli olabilir.
Axenfeld-Rieger Sendromu Belirtileri
Bu liste, bu hastalığa sahip kişilerin sahip olabileceği semptomları listeler. Çoğu hastalık için semptomlar kişiden kişiye değişecektir. Aynı hastalığı olan kişiler, listelenen tüm semptomlara sahip olmayabilir.
- Az gelişmiş iris
- Küçük bir kornea (mikrokornea)
- Korneanın dış kenarı etrafında opak bir halka
- Gözün önünde yapışıklıklar
- Merkez dışı bir göz bebeği (korektopi)
- Fazladan gözbebeği (polikori)
- Göz içi basınçta artış (glokom)
- Normalden daha az dişin olması (hipodonti)
- Normalden daha küçük bir diş veya dişler (mikrodonti) ve / veya koni şeklindeki dişler
- Geniş ve düz bir burun köprüsü, çıkıntılı alt dudak ve alın, geniş aralıklı gözler ve basık bir yüz
- Üst çenenin yetersiz gelişmiş kemikleri (hipoplazi)
- Alışılmadık derecede küçük bir anal açıklık (anal stenoz)
- Göbek çevresindeki derinin doğumdan sonra küçülmemesi
- Göbek fıtığı veya göbek çevresindeki karın duvarındaki bir zayıflık nedeniyle bağırsakta çıkıntı
- İdrar deliğinin penisin alt tarafında açılması (hipospadias)
Nasıl Teşhis Edilir?
Bozukluk genellikle karakteristik göz kusurlarının varlığıyla tanınır. Bazı durumlarda bunlar gecikir ve tanı, küçük konik dişlerin ortaya çıktığı erken çocukluk dönemine kadar ertelenebilir.
Axenfeld-Rieger Sendromu Tedavisi
Rieger sendromunun tedavisi semptomatik ve destekleyicidir. İlaç tedavisi, genellikle göz damlalarından oluşan glokom için birincil tedavidir. Lazer cerrahisi genellikle göz içindeki basıncın ilaçlarla giderilemediği hastalar için ayrılmıştır. Ameliyat gerekliyse tercih edilen prosedür trabekülektomidir.
Korektopi ve polikori olan hastalar ışığa çok fazla duyarlılık (fotofobi) yaşayabilir, bu hastalara kontakt lensler yardımcı olabilir.
Kişinin ek bulguları varsa, tedavi mevcut semptomlara bağlı olacaktır ve yüz veya diş problemlerini düzeltmek için ameliyat, kalp ameliyatı veya hipospadias vakaları için düzeltici ameliyatı içerebilir. Büyüme hormonu eksikliğine bağlı kısa boy, büyüme hormonu ile tedavi edilebilir.
Bu Hastalık İçin Kullanılan Diğer İsimler
- ARS
- Axenfeld ve Rieger anomalisi
- Axenfeld anomalisi
- Axenfeld sendromu
- AXRA
- AXRS
- Rieger anomalisi
- Rieger sendromu
Kaynaklar ve Referanslar:
1: Axenfeld-Rieger syndrome
2: About Axenfeld-Rieger syndrome
3: What is axenfeld rieger syndrome?