Oftalmo-Akromelik Sendrom Nedir? Belirtileri, Nedenleri, Tedavisi
Oftalmo-akromelik sendrom (ophthalmo-acromelic syndrome), gözlerin, ellerin ve ayakların malformasyonlarına (kusurlarına) neden olan nadir bir genetik bozukluktur. Bu durumun özellikleri doğumdan itibaren mevcuttur.
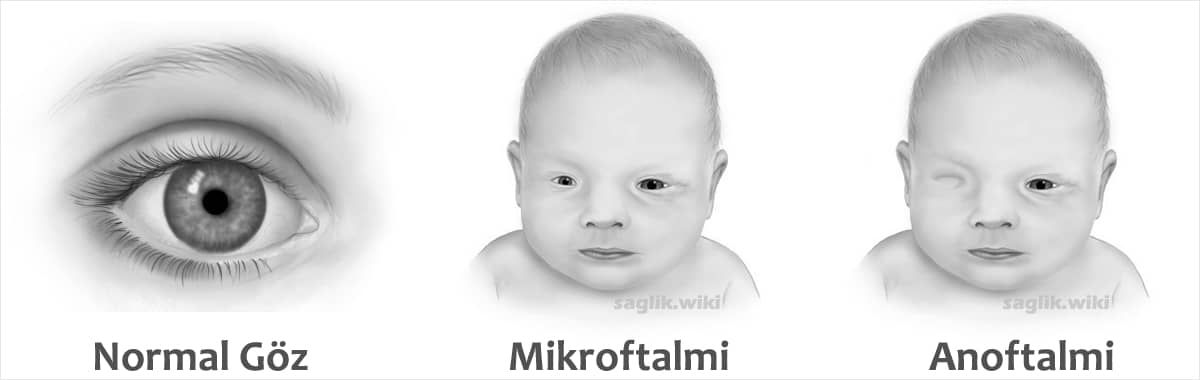
Gözler genellikle yoktur veya çok az gelişmiştir (anoftalmi) veya anormal derecede küçük olabilirler (mikroftalmi). Bu durumda genellikle her iki göz de benzer şekilde etkilenir, ancak sadece bir göz küçükse veya eksikse, diğer gözün yapılarında boşluk veya yarık gibi bir kusur olabilir (kolobom).
Oftalmo-akromelik sendromda en sık görülen el ve ayak malformasyonu parmak veya ayak parmaklarının eksikliğidir (oligodaktili). Diğer sık görülen malformasyonlar arasında birbirine kaynaşmış parmaklar veya ayak parmakları (sindaktili) veya ekstra parmaklar veya ayak parmakları (polidaktili) bulunur.
Bu iskelet malformasyonları genellikle akromelik olarak tanımlanır, yani vücudun merkezinden uzaktaki kemiklerde meydana gelir.
Kolların ve bacakların uzun kemiklerini veya omurga kemiklerini (omurlar) içeren ek iskelet anormallikleri de ortaya çıkabilir.
Bu durumun ek özellikleri arasında ağzın çatısında açıklık (yarık damak), dudakta bir açıklık (yarık dudak), benzersiz yüz özellikleri ve öğrenme sorunları (zihinsel engeller) yer alabilir.
Nadir durumlarda at nalı böbrek, venöz ve vertebral anomaliler de bildirilmiştir. Birkaç vakada postnatal / perinatal ölüm meydana gelmiştir.
Oftalmo-Akromelik Sendrom Ne Kadar Yaygındır?
Oftalmo-akromelik sendromun prevalansı (görülme sıklığı) bilinmemektedir, ancak bugüne kadar çoğunlukla akraba olan ebeveynlerden olmak üzere 35’ten fazla vaka bildirilmiştir.
Oftalmo-Akromelik Sendromun Nedenleri
Oftalmo-akromelik sendrom, SMOC1 genindeki bir değişiklikten (mutasyon) kaynaklanır. SMOC1 geni, salgılanan modüler kalsiyum bağlayıcı protein 1 (SMOC-1) adı verilen bir proteini yapmak için talimatlar sağlar.
Bu protein, birçok dokudaki hücreleri destekleyen ve embriyonik gelişim sırasında hücreleri birbirine tutturmaya yardımcı olan ince, tabaka benzeri yapılar olan bazal zarlarda bulunur.
SMOC-1 proteini, birçok farklı proteine bağlanır ve vücuttaki dokuların büyümesini ve gelişmesini uyaran büyüme faktörleri adı verilen molekülleri düzenlediği düşünülmektedir.
Bu büyüme faktörleri, iskelet oluşumunda, uzuvların normal şekillenmesinde (modellemesinde), göz oluşumu ve gelişiminde önemli rol oynar. SMOC-1 proteini ayrıca osteoblastlar olarak adlandırılan kemik oluşturan hücrelerin olgunlaşmasını (farklılaşmasını) da teşvik eder.
SMOC1 gen mutasyonları genellikle işlevsel olmayan bir SMOC-1 proteini ile sonuçlanır. SMOC-1’in kaybı, iskeletin, uzuvların ve gözlerin normal gelişimini bozacak şekilde büyüme faktörü sinyalini bozabilir.
Bu değişiklikler muhtemelen oftalmo-akromelik sendromun anoftalmi ve iskelet malformasyonlarının temelini oluşturmaktadır. SMOC1 gen mutasyonlarının bu durumun diğer özelliklerine nasıl yol açtığı belirsizdir.
Oftalmo-akromelik sendromlu bazı kişilerin SMOC1 geninde tanımlanmış bir mutasyonu yoktur. Bu kişilerde durumun nedeni bilinmemektedir.
Oftalmo-Akromelik Sendrom Nasıl Kalıtılır?
Bu durum otozomal resesif bir şekilde kalıtsaldır. Bu, kişinin bu duruma sahip olması için sorumlu genin her iki kopyasında da (her bir ebeveynden değişmiş bir kopya) bir değişikliğe (mutasyona) sahip olması gerektiği anlamına gelir.
Otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri mutasyona uğramış genin bir kopyasını taşır ve taşıyıcı olarak adlandırılır. Taşıyıcılar tipik olarak durumun belirti ve semptomlarını göstermezler.
Otozomal resesif bir durumun iki taşıyıcısı çocuk sahibi olduğunda, her çocuğun bu duruma sahip olma riski % 25 (4’te 1), ebeveynlerin her biri gibi bir taşıyıcı olma riski %50 (2’de 1) ve koşula sahip olmama ve taşıyıcı olmama şansı %25’tir. Bu sonuçlar rastgele (şans eseri) meydana gelirler.
Oftalmo-Akromelik Sendrom Belirtileri
Belirtiler konjenitaldir yani doğumdan itibaren mevcuttur ve şunları içerebilir:
- Bir veya iki gözün alışılmadık şekilde küçük olması (mikroftalmi)
- Bir veya iki gözün yokluğu (anoftalmi)
- Göz yapılarında boşluk veya yarık (koloboma)
- El ve ayak parmaklarının eksikliği (oligodaktili)
- Birbirine kaynaşmış parmak veya ayak parmakları (sindaktili)
- Ekstra parmak veya ayak parmakları (polidaktili)
- Uzun kemik hipoplazisi (kusurlu veya eksik gelişim)
- Yarık damak ve yarık dudak
- At nalı böbrek
- Basık bir orta yüz
- Seyrek kirpikler
- Kısa palpebral fissürler (alt ve üst göz kapağı arasındaki açıklık)
Nasıl Teşhis Edilir?
Tanı, karakteristik klinik bulguların varlığına dayanır. Bilgisayarlı tomografi (BT) taramaları ve manyetik rezonans görüntüleme (MRG), küre, optik sinir ve ekstra oküler kasların varlığını veya yokluğunu belirlemede yardımcı olabilir. Ultrason da hastalıkla ilişkili uzuv anomalilerini tanımlamak için kullanılabilir.
SMOC1 genindeki bir mutasyonun belirlenmesi tanıyı doğrular. Bir ailede nedensel mutasyon tespit edilmişse, CVS veya amniyosentez yoluyla doğum öncesi test mümkündür.
Oftalmo-Akromelik Sendrom Tedavisi
Oftalmo-akromelik sendromun tedavi yoktur. Anoftalmi / mikroftalmi tedavisi, bir oküloplastik cerrah ve göz doktoru ile tartışılabilir.
Anoftalmi için, doğumdan sonra mümkün olan en kısa sürede göz kapaklarının, yuva ve orbital kemiklerin genişletilmesi önerilir. Bu, bir göz doktoru tarafından konformer tedavisi veya hidrojel soket genişleticiler kullanılarak oküloplastik cerrahi ve ardından orbital implantlar veya dermis-yağ greftleri ile yapılır.
Bu işlemin uygulanması, hastaların yüz deformitesini önleyerek daha tipik bir görünüm elde etmelerine yardımcı olabilir. Biraz görmesi olanlar (mikroftalmi şiddetli değilse) görsel araçlardan yararlanabilir.
Hastanın hareketlilik veya işlev kazanmasına yardımcı olmak için bazı uzuv anormallikleri de cerrahi olarak düzeltilebilir, bu nedenle ortopedik değerlendirme gereklidir.
Bu hastalığa sahip tüm bireyler değerlendirilmeli ve özel eğitim gerekli olabilir.
Bu Hastalık İçin Kullanılan Diğer İsimler
- Anoftalmi-sindaktili
- Anoftalmi-Waardenburg sendromu
- Uzuv anomalili anoftalmi
- Anoftalmi-uzuv anomalileri sendromu
- Uzuv anomalili mikroftalmi
- OAS
- Oftalmokromelik sendrom
- Sindaktili-anoftalmi sendromu
- Waardenburg anoftalmi sendromu
Kaynaklar ve Referanslar:
1: Anophthalmos with limb anomalies
2: About ophthalmo-acromelic syndrome
3: Ophthalmo-acromelic syndrome (OAS)