Akondrogenezis Nedir? Belirtileri, Nedenleri, Tedavisi
Akondrogenezis (achondrogenesis), kıkırdak ve kemik gelişimini etkileyen ciddi bir hastalık (iskelet displazisi) grubudur. Akondrogenezis (akondrogenezis sendromu) küçük bir beden, dar göğüs, kısa ekstremite (kollar, bacaklar) ve diğer iskelet anomalileri ile karakterize adilen nadir ve şiddetli bir genetik bozukluk grubudur. Vücutta yeterli büyüme hormonu üretilmez ise sonuç olarak kıkırdak ve kemik düzgün gelişim göstermez bu da iskelet sistemi anormalliklerine neden olur. Hastalığın üç türü vardır, bunlardan tip 1A ve tip 1B otozomal resesif (çekinik) geçiş bozuklukları iken tip2 otozomal dominant (baskın) bir hastalıktır. Her her üç tipte genellikle gebeliğin 14-17. haftalarında doğum öncesi muayene ile saptanabilir.
 Bu nadir hastalık kalıtsaldır. Hem baba hemde anne hatalı geni taşıyorsa bu bozukluğu olan bir çocuğa sahip olma ihtimali daha yüksektir. Genetik bozukluk resesif olduğundan genin ebeveyn taşıyıcıları durumun herhangi bir belirtisine işaret etmez. Akondrogenezisli bebeklerin çoğu doğumdan önce veya sonra düzgün bir şekilde nefes alamadıkları için hayatlarını kaybetmektedirler. Bununla birlikte bazı bebekler yoğun tıbbi destekle kısa süre yaşamışlardır. Bozukluk için şuanda mevcut hiçbir tedavi yoktur.
Bu nadir hastalık kalıtsaldır. Hem baba hemde anne hatalı geni taşıyorsa bu bozukluğu olan bir çocuğa sahip olma ihtimali daha yüksektir. Genetik bozukluk resesif olduğundan genin ebeveyn taşıyıcıları durumun herhangi bir belirtisine işaret etmez. Akondrogenezisli bebeklerin çoğu doğumdan önce veya sonra düzgün bir şekilde nefes alamadıkları için hayatlarını kaybetmektedirler. Bununla birlikte bazı bebekler yoğun tıbbi destekle kısa süre yaşamışlardır. Bozukluk için şuanda mevcut hiçbir tedavi yoktur.
Bilgicik: Akondrogenezisin 1A ve 1B türleri nadir görülen genetik bozukluklardır ve insidansı bilinmemektedir. Kombine olarak Tip 2 ve hipokondrojenez (benzer bir iskelet bozukluğu) 40.000 ila 60.000 yenidoğanda 1 görülür.
Akondrogenezisin Türleri
Bu bozukluk genellikle kalıtım paterni, belirtiler ve semptomlara dayalı olarak üç farklı türe ayrılır.
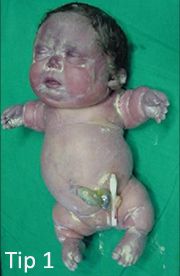
1A Tipi (Houston-Harris)
Akondrogenezis tip 1A’da bebeğin kafatası kemiği çok yumuşaktır ve uzuvlar çok kısadır. Her üç tipte olduğu gibi göğüs dar olduğundan bebek zor nefes alır. Omurga ve pelvis kemikleri iyi şekillenmemiştir ve bebeğin kaburgaları kısa ve kırılgan olur. İki taşıyıcı ebeveynin etkilenen bir çocuğu olma riski her gebeliğin % 25’idir. Her gebelikte ebeveyn gibi taşıyıcı olan bir çocuğa sahip olma riski % 50’dir. Bir çocuğun her iki ebeveynden de normal gen alması şansı % 25’dir.
1B Tipi (Parenti-Fraccaro)
Tip 1B, 1A’ya çok benzerdir ve iki tip arasındaki ayırımı genellikle genetik bir test gerektirir. Çok kısa ekstremiteler ve dar göğüslere ek olarak 1B tipinde yuvarlak göbek ve bazen kasık veya göbek düğmesine yakın bir kese bulunur. Bebeğiniz bu tür akondrogenezise sahipse ayakları içe doğru döndük olabilir. Ayrıca bu tipte ayak ve parmaklar da çok kısadır.
Tip 2 (Langer-Saldino)
Diğer iki tipte olduğu gibi tip 2 ile doğan bebekler kısa ekstremiteler ve dar bir göğüse sahip olurlar. Kaburgaları genellikle kısadır ve ciğerleri, omurgası ve pelvis kemikleri gelişmemiştir. Ağız çatısı yarık damak olarak bilinen bir açıklığa sahip olabilir. Göbek büyük olabilir ve doğumdan önce bazı bebekler bedenlerinde çok fazla sıvıya sahip olurlar. Ayrıca küçük bir çene ve alın diğer belirtilerdir. Anormal genden etkilenen ebeveynden bebeğe geçme riski her gebelik için % 50’dir.
Akondrogenezis Nedenleri Nelerdir?
Hastalığın tüm tipleri belirli bir genin mutasyonundan kaynaklanır. Genler vücudun birçok fonksiyonunda kritik bir rol oynayan proteinlerin oluşturulması için talimatlar sağlarlar. Bir genin bir mutasyonu oluştuğunda protein ürünü hatalı, yetersiz veya eksik olabilir. Belirli proteinin işlevlerine bağlı olarak bu vücudun birçok organ sistemini etkileyebilir. Akondrogenezis tip 1A ve tip 1B otozomal resesif kalıtım kalıbına sahiptir bu da her hücredeki TRIP11 veya SLC26A2 geninin her iki kopyasında mutasyona neden olduğu anlamına gelir. Çoğu zaman otozomal resesif bir durumu olan bir bireyin ebeveynleri mutasyona uğramış genin bir kopyasını taşır ancak durumun bulgu ve belirtilerini göstermezler.
Akondrojenez tip 2 ise otozomal dominant bir bozukluk olarak düşünülür çünkü her bir hücredeki değiştirilmiş genin bir kopyası bu duruma neden olabilir. Hemen hemen her zaman COL2A1 genindeki yeni mutasyonlardan kaynaklanır ve genellikle ailelerinde bozukluk öyküsü olmayan insanlarda görülür.
Akondrogenezisin Belirtileri Nelerdir?
Kısa ekstremiteler ve dar bir göğüs tüm türler için ortak belirtilerdir. Diğer semptomlar türüne göre değişir ve tam türünü ayırt etmek için genetik test gerekebilir.
1A Tipi Belirtileri
- Yumuşak kafatası kemiği
- İyi oluşmamış omurga ve pelvik kemikler
- Kısa ve kolayca kırılabilir kaburgalar
1B Tipi Belirtiler
- Yuvarlak göbek
- Göbek yakınında torba
- Kısa ayak ve parmaklar
- İçe dönük ayaklar
Tip 2 Belirtiler
- Kısa kaburga
- Kötü gelişmiş akciğerler
- Yarık dudak (tavşan dudak)
- Küçük çene yapısı
- Büyük alın ve göbek
Teşhis Nasıl Yapılır?
Fiziksel özellikler, röntgen (radyografik) bulgular ve doku örneklerinin mikroskopta incelenmesi (histoloji) ile hastalık teşhis edilebilir. Teşhisi doğrulamak için biyokimyasal testler ve moleküler genetik testler de kullanılabilir.
Gebeliğin 14-15 haftasından sonra hastalığın ultrasonla prenatal tanısı mümkündür. Spesifik gen mutasyonları bir aile üyesinde tanımlandıysa koryonik villüs örneklemesi (gebeliğin 10-12. haftası) veya amniyosentez (gebeliğin 15-18. haftası) ile doğum öncesi tanı mümkündür.
Bilgicik: Akondrogenezis terimi ilk kez 1952 yılında Marco Fraccaro adlı bir İtalyan patolog tarafından kullanılmıştır. Akondrogenez terimi Yunanca’dan türetilmiştir ve “kıkırdak üretmemek” anlamına gelir.
Akondrogenezis Tedavisi
Hastalık için hiçbir tedavi yoktur. Uygulanan tedaviler sadece destekleyicidir.
Hastalık ailenizde mevcutsa bozukluğun önlenmesi için tek seçenek bir genetik danışmanlığa sahip olmanızdır. Genetik danışma sırasında bir uzman tıbbi geçmişinizi toplayacak ve ailelerinizin her iki tarafında meydana gelen hastalıkları soracaktır. Doktorlar siz ve eşinizin hastalığın oluşmasında etken olan genleri taşıdığınızı belirlemek için testler yapacaktır. Çiftler bebek sahibi olmak için karar vermeden önce bu seçeneği kullanabilir.